
A clinical trial is a type of research that studies a test or treatment given to people. Clinical trials study how safe and helpful tests and treatments are. When found to be safe and helpful, they may become tomorrow’s standard of care.
Clinical trials are experiments or observations done in clinical research. Such prospective biomedical or behavioral research studies on human participants are designed to answer specific questions about biomedical or behavioral interventions, including new treatments (such as novel vaccines, drugs, dietary choices, dietary supplements, and medical devices) and known interventions that warrant further study and comparison. Clinical trials generate data on safety and efficacy.They are conducted only after they have received health authority/ethics committeeapproval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial – their approval does not mean that the therapy is ‘safe’ or effective, only that the trial may be conducted.
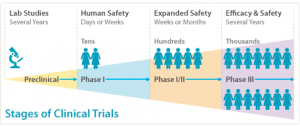
Depending on product type and development stage, investigators initially enroll volunteers or patients into small pilot studies, and subsequently conduct progressively larger scale comparative studies. Clinical trials can vary in size and cost, and they can involve a single research center or multiple centers, in one country or in multiple countries. Clinical study design aims to ensure the scientific validity and reproducibility of the results.
Costs for clinical trials can range into the billions of dollars per approved drug. The sponsor may be a governmental organization or a pharmaceutical, biotechnology or medical devicecompany. Certain functions necessary to the trial, such as monitoring and lab work, may be managed by an outsourced partner, such as a contract research organization or a central laboratory.
Only 10 percent of all drugs started in human clinical trials become an approved drug.
Medical research may be broadly defined as any type of study or experiment performed for the purpose of producing generalizable knowledge related to human health or medical treatment. Medical Research performed on Human Beings could involve observation or some form of intervention (whether physical, chemical, and psychological). Medical Research performed on patients will be described as ‘Clinical Research’. Not all medical research is Clinical Research, so defined. In fact, new pharmaceuticals are often first tested on animals and healthy human volunteers, before they are tested on patients’ volunteers. Not all clinical research requires direct contact with the patients. Epidemiological research, involving the study of health related condition in population, rarely requires direct contact .
This dissertation tries to probe into the modern practice of referring to those upon as ‘participants’. Many regulatory instruments use the term ‘Subject’ instead of ‘Participant’. Many commentators prefer ‘participant’, as a less passive term to emphasize the need to ensure that person’s consent or, if he lacks capacity, at least acquiescence. Both terms have the potential to create misunderstandings; ‘participant’ might be mistakenly considered to refer to the researcher and ‘subject’ might be mistakenly referred to the thing (such as new therapy or drug) being tested. Terminological consistency is the best way of avoiding the misunderstanding .
INNOVATIVE TREATMENT is a feature of medical care, as doctors at times modify their medical practices in the light of what they learn from patient’s experiences and this will sometimes lead to experimentation with a new untested therapy . This is just one reason why it is not always easy to distinguish between research and treatment. The primary aim of medical research is to produce new knowledge for the benefit of future patients. The primary aim of medical treatment is to benefit the immediate patient. Activities can, however have multiple aims and it is possible to mislead and be misled. A doctor who uses an unproven therapy could intend to benefit future patients as a primary or a secondary plan.
The Principal of Personal Care
“The traditional concept of the physicians’ relation to his patient is one of unqualified fidelity to that patient’s health “. Schafer in1982 noted that, in his traditional role of healer, the physician’s commitment is “exclusively and unequivocally to promote the interests of his patient.” This concept has no accepted name; it is very closely related to what was called the “Hippocratic principle” and has also been named the “therapeutic obligation.” It relates specifically to the professional relationship between physician and patient and expresses the physician’s obligation “to place the patient’s interests before all else within the professional relationship.”
The personal care principle is commonly stated as the physician’s duty “to do what is best for the patient.” It is not entirely clear whether it is implied to the Hippocratic Oath (“Whatever houses I may visit, I will come for the benefit of the sick”), but the Physician’s Oath adopted by the World Medical Association at Geneva (1948) is explicit: “the health of my patient will be my first consideration.” In the context of clinical trials, the principle appears in the Medical Research Council of Great Britain’s (1964) statement on Responsibility in Investigations on Human Subjects: “It goes without question that any doctor taking part in a collective controlled trial is under an obligation to withdraw a patient from the trial, and to institute any treatment he considers necessary, should this, in his personal opinion, be in the better interests of his patient.” And individual, authors writing about clinical trials routinely acknowledge ” the fundamental principle that the physician, investigator’s primary responsibility is to his patient” .
In addition to whatever moral force might support the personal care principle, there is a practical need for it; its absence would create severe obstacles to the proper functioning of the present system of medical care. One who doubts that his physician’s professional advice is motivated by what is best for the patient is effectively cut off from adequate care. Blind faith that no physician ever put another interest above the health of a patient is not necessary or even desirable. But confidence that the personal care principle is usually honoured seems essential if people are to be motivated to seek out needed medical help and to follow prescribed therapies .
Views about the proper relationship between physician and patient are not static. Before the 1940s the dominant image was a paternalistic one in which the focus was on the physician’s responsibility to do what, in his judgment, was best for the patient. Recent decades have seen increasing emphasis on the rights of the autonomous patient, particularly the rights to be informed and to make his or her own decisions. The personal care principle does not demand that the physician does whatever he thinks is best for the patient, regardless of the patient’s preferences: the role of the physician is to advise and propose, not to impose his judgments. The right to decide what is truly in the patient’s best interests belongs to the patient. As Chapman (1984) put it “There are times when the doctor does indeed know best, even though he or she may not impose a decision; ultimate choice is incontestably the patient’s.” This view is exempli- fied in recent legal judgments that the decision of a patient whose religious beliefs forbid blood transfusion must be respected, even by a physician who is convinced of the urgent medical need for the transfusion. Of course many patients choose to play a passive role, trusting the physician to do “Whatever you think is best.” And of course the basis for this decision is the assumption that the personal care principle will be respected.
Competence
The personal care principle is not the physician’s only rule of professional ethics. Another obligation relates directly to our discussion: the physician must maintain competence. His actions are ethically sound only if they are directed by both the personal care principle and adequate professional knowledge and skills. A physician does not face an obligation to provide the best available therapy for his patient. His obligation is to provide what is best in his judgment, and to ensure that his judgment is medically competent. No more is humanly possible. If two competent clinicians, both observing the personal care principle, disagree about the relative merits of two therapies, then one may be using what is in fact an inferior treatment, but neither is behaving unethically.
Limitation to Personal care
There are certain situations in which the physician’s responsibility to serve the interests of the patient is restricted on behalf of the interests of society. For example, although the physician is generally charged with maintaining confidentiality, he is required to report gunshot wounds to the police. Clearly such a report might be contrary to the interests of a wounded fugitive. For another example, Food and Drug Administration regulations limit the conditions under which many drugs can be used, so that a physician may not be allowed to prescribe what he or she thinks would be best for the individual patient. It is important to note that these situations, in which the personal care principle is compromised for a presumed social good, are explicitly circumscribed by public law and not left to the personal judgment of the physician .
THE PROBLEM:
After the liberalization of Clinical Trial procedure by making amendment to schedule Y of the drug and cosmetics act it has become easy to go for phase II type of clinical trials which are elementary trials with no clear success history, hence becoming a danger for the subjects to the trials. The First Research problem tries to identify the problem of uninformed consent and the lack of knowledge of legal rights to compensation for the people who volunteer for being a subject to these Clinical Trials in India.
The Second Research problem tries to bring up the issue of the lack of clear legal backing for the humongous amounts of clinical Trials conducted in India with no strict laws to supervise and support.
OBJECTIVES OF THE STUDY:
The primary objective to study in the given area of Clinical Trial agreements or Human experimentation is the idea of it being whether or not an ethical means to better technology, whether it is correct to use healthy human beings or even the one with a terminal disease as a means to an end for the sake of better productivity and technological advancement in the medical field just to provide a better medi-care and health facility to the upcoming future generations.
The secondary objective to research in the given area is to probe in to the facts as to whether how advanced is the Indian law in comparison to other countries law as to handle the increasing clinical trial agreements and the lack of such strong back end of law leading to unauthorized unregistered Human experimentation.
METHODOLOGY
The methodology to be used in this dissertation is doctrinal in nature including Secondary Sources that include books, journals, reports and articles available online.
IMPORTANCE OF STUDY
India has a huge number of patients who can help to contribute to the advancement of medical sciences. By using India, the multinational pharma companies were hoping to tap into this rich resource and to fast forward their turn-round time for the trials; but this should not mean any compromise on safety standards. There has been a constant lack of research data and discussion on this subject and many a times the people who are treated as scape goats have no knowledge of what they are going to go through hence there needs to be serious research on this topic to pass on the message wide and clear.
CHAPTER 2 : LITERATURE REVIEW
ARTICLES:
Richard Singer, Consent of the Unfree: Medical Experimentation and Behavior Modification in the Closed Institution. Law and Human Behavior, Vol. 1, 1977, 101-162
This article talks about the problem of free consent and free will. Moreover this article talks about the rate of volunteerism and how many people are forced to be a part of such clinical trials without free will and further on talks about coercion or consent through inducement for clinical trials.
Hans Jonas, Beyond Voluntary Consent: Hans Jonas on the Moral Requirements of Human Experimentation Journal of Medical Ethics, Vol. 19,1993, 99-103
In this article Hans Jonas contends that except in cases of widespread medical emergencies, people do not have a moral or social obligations to volunteer to be subjects in medical experiments. The author further argues that any appeal for volunteer subjects in medical experiments should whenever possible give priority to those who can identify with the project and offer a strong sense of commitment to its goals.
Richard M. Royall , Ethics and Statistics in Randomized Clinical Trials ,Institute of mathematical Statistics Science, Vol. 6, No. 1 1991, pp. 52-62
This article talks about Randomized clinical trials and how this scheme of clinical trials is better than many other schemes often practiced in the medical world. This article taks about the relationship of the patient with the doctor and through these responsibilities how he is responsible for the health of the doctor.
Sandhya Srinivasan , The Clinical Trials Scenario in India Economic and Political Weekly, Vol. 44, 2009, pp. 29-33
The Indian government has in the recent years promoted India as a fast upcoming Clinical trial destination. This article talks about the regulation and changes in the enactment that has been made by the Indian government leading to easy access of clinical trials for research organization.
Andrew Herxheimer , Clinical Trials: Two Neglected Ethical Issues Journal of Medical Ethics, Vol. 19, No. 4 (Dec., 1993), pp. 211, 218
This article has discussed two very important issues(1) that a proposal for a clinical trial should be accompanied by a thorough review of all previous trials that have examined the same and closely related problems. (2) that a trial should be approved by a research ethics committee only if the investigator undertakes to register it in an appropriate register of clinical trials as soon as one exists.
BOOKS
Shaun D. Pattinson, Medical Law and Ethics , Published by Sweet & Maxwell
This Book has a chapter named Medical Research which discusses about the history of medical trials, the Helsinki code, and then further moves on To discusses about Controlled trials and the importance of consent for volunteering to be a subject in the Trials.
Emily Jackson, Medical Law Text, Cases, and Materials, Published by Oxford University Press.
This book has a chapter named Clinical Research talks about animal experimentation and then the International codes that regulates the clinical Trials.
Dr. Lily Srivastava, Law & Medicine Published by Universal Law publishingCo. Pvt. Ltd.
This book has a chapter on Clinical Trails which more than just discussing about the history , evolutions and legal aspects of clinical trials , this chapter also dicusses the cases that have been brought up for non-ethical Trials.

Chapter 3 : HISTORY AND EVOLUTION
The two ancient Indian scripts Charkha Samhita (a text on medicine) and Sushruta Samhita (a text book of surgery), complied as early as 200 B.C and 200A.D. respectively shows that medical research is not a new concept for India. However, a lot has changed in the Clinical research scenario since then.
The world’s first clinical trial was recorded in the ‘Book of Daniel’ in The Bible. This experiment resembling a clinical trial was not conducted by a medical expert, but by King Nebuchadnezzar a resourceful military leader. During his rule in Babylon, Nebuchadnezzar ordered his people to eat only meat and drink only wine, a diet he believed would keep them in sound physical condition. But several young men of royal blood, who preferred to eat vegetables, objected. The king allowed these rebels to follow a diet of legumes and water but only for 10 days. When Nebuchadnezzar’s experiment ended, the vegetarians appeared better nourished than the meat-eaters, so the king permitted the legume eaters to continue their diet. This probably was the one of the first times in evolution of human species that an open uncontrolled human experiment guided a decision about public health .
Avicenna (1025 AD) in his encyclopedic ‘Canon of Medicine’ describes some interesting rules for the testing of drugs. He suggested that in the clinical trial a remedy should be used in its natural state in disease without complications. He recommended that two cases of contrary types be studied and that study be made of the time of action and of the reproducibility of the effects. These rules suggest a contemporary approach for clinical trials. However, there seems to be no record of the application of these principles in practice.
The first clinical trial of a novel therapy was conducted accidentally by the famous surgeon Ambroise Pare in 1537. In 1537 while serving with the Mareschal de Motegni he was responsible for the treatment of the battlefield wounded soldiers. As the number of wounded was high and the supply of conventional treatment oil was not adequate to treat all the wounded soldiers, he had to resort to unconventional treatment. He describes,’ at length my oil lacked and I had to apply in its place a digestive made of yolks of eggs, oil of roses and turpentine. That night I could not sleep at any ease, fearing that by lack of cauterization I would find the wounded upon which I had not used the said oil dead from the poison. I raised myself early to visit them, when beyond my hope I found those to whom I had applied the digestive medicament feeling but little pain, their wounds neither swollen nor inflamed, and having slept through the night. The others to whom I had applied the boiling oil were feverish with much pain and swelling about their wounds. Then I determined never again to burn thus so cruelly the poor wounded by harquebuses’. However, it would have taken another 200 years before a planned controlled trial would be organized.
The Hippocratic ethical principle of primumnon nocere is a time- tested principle that has governed the field of medicine , but an international discourse on the subject was wanting since before the Second World War . One would argue, given the extensive medical experimentation that occurred in that era, especially in the Nazi regime, that the existence of laws, guidelines or regulations should have been a condition precedent. The lack of any such legal or ethical regulation, however, led to disregard for the sanctity of human life with physicians advocating the idea of ‘State before the individual’ , which is characteristic of the Nazi era. Human beings who served as clinical trial subjects were at the mercy of the physicians, who made no distinction in the treatment doled out to lab rats and humans. In responding to the ‘national threat’ of disease, physicians acted in contravention to their ethical obligations .
The Separation of ethics from the field of medicine spurred the international medical community into action. Various international declarations and codes emerged to regulate medical experimentation. The instrumental ones are discussed hereinafter.
THE NUREMBERG CODE
The Nuremberg Code (‘the Code’) was developed by the American judges who adjudicated the Nuremberg Trial . It involved the convergence of Hippocratic ethics and the protection of human rights into a single code . The ten principles articulated were constructed with the research subject’s welfare as the focal point. This implied a change in perception with regard to the status of the research subject. While the Hippocratic ethics viewed the subject as ‘passive and dutiful’ and assumed that the physician knows best, they nevertheless imposed ethical obligations on the physician to prioritize the welfare of the subject. The Code recognized the autonomy of an individual, at the core of which is the codification of ‘Consent’. In according positive rights to the subjects, it destabilized the notion of passivity.
In the Code, the principle reflecting the recognition of individual autonomy is that of voluntary consent. In order to effectuate individual autonomy, Principle 1 of the Code provides that consent is a non-negotiable precedent to an individual’s participation in a clinical trial . The implications of the same are threefold. Firstly, a participant should have the legal capacity to give consent. Secondly, such consent should be voluntary and free, and not a by-product of force, coercion, fraud, deceit, duress, etc. Thirdly, such consent should be an exercise of informed choice, involving ‘sufficient knowledge and comprehension about the subject-matter’ . The individual conducting the experiment ought to discern the quality of the consent. The remainder of the provisions deal with facets of clinical trial, the design of the experiment, expected outcomes and risk mitigation, prohibiting an experiment wherever a strong likelihood of disability or death exists, adequacy of preparation, facilities, the quality of risk control equipment and the obligation of the researcher to terminate the experiment if required.
The Code has not acquired the status of binding international law but it has nevertheless been instrumental in shaping medical ethics and serves as a model law . Critics of the Code claim that it is a copy of the Guidelines for Human Experimentation, 1931, and that the lack of acknowledgment to that effect amounts to plagiarism. Despite the divided schools of thought on the subject of the Code, even its critics cannot gainsay that this Code catalyzed the shift in approach with respect to the status of the research subjects, restoring the dignity they deserve and conforming to their autonomy .
THE DECLARATION OF HELENSIKI
A document confirming the rights of the subject would be rendered ineffectual, if measures to check the excesses of the physician/researcher were not supplanted. The Declaration of Helsinki (‘the Declaration’) expounded on the obligations of the physician/researcher. A draft of the Declaration was tabled in 1961, and after subjecting it to intensive examination, it was adopted as the Declaration of Helsinki in1964 . The Declaration went through six amendments , and the version in use currently is the one amended in 2008.
The salient provisions of the Declaration are classified into three categories: the principles concerned with the obligation of the physician, the principles of consent, and the principles advocating transparency to bring about the effective regulation of clinical trials .
It is worth noting that the established concept of consent undergoes an evolution in the Declaration. It goes a step further by recognizing that groups lacking capacity to consent are vulnerable. Principle 17 is one such medium for providing this protection. It excludes these vulnerable groups from being used as subjects unless the research is in furtherance of their interest, so that their lack of capacity is not exploited to further research in rem. As a further check against exploitation, it mandates that the consent of the subject’s legal representative has to be supplanted by the assent of the subject wherever possible .
The principles of the Declaration have been emulated through implicit and explicit incorporation and are accepted as a uniform standard of ethics applicable to trials The Declaration finds place in national legislation, ordinances and references as well. A noteworthy example is that of Uganda, a country which in spite of being a developing country, has incorporated the Declaration as well as the Code in its 1997 Guidelines for the Conduct of Health Research Involving Human Subjects .
CIOMS GUIDELINES ‘ INTERNATIONAL ETHICAL GUIDELINES FOR BIOMEDICAL RESEARCH INVOLVING HUMAN SUBJECTS
The challenges faced by the declaration, despite the recognition of its authority in the world for a century created doubts about its acceptance under the social political and cultural framework and with the Clinical Trials increasing at a constant ratio the principles set forth in the Declaration were rendered ineffectual when applied to outsourced clinical trials in less developed countries. This was either because the developed countries would apply these principles with complete disregard to the culture, socio-economic scenario and legal infrastructure of the less developed country, or would not apply the principles at all.
It was to prevent such instances as occurred in the developing countries that the ‘Council for International Organizations of Medical Sciences’ (‘CIOMS’) in collaboration with the World Health Organization prepared guidelines to indicate how the ethical principles to guide the conduct of biomedical research involving human subjects set forth in the Declaration could be effectively applied in developing countries. The CIOMS guidelines thus served as the key to customize the Declaration as suited to different circumstances, while the basic structure remained untouched. These guidelines were the 1982 version of Proposed International Ethical Guidelines for Biomedical Research Involving Human Subjects. In time, these were revised and bifurcated into the International Guidelines for Ethical Review of Epidemiological Studies, 1991, and the International Ethical Guidelines for Biomedical Research Involving Human Subjects, 1993 .
CONCLUSION
The third chapter of this dissertation i.e. History and Evolution starts by mentioning that the clinical trials in india were first mentioned in the Vedas, i.e. Charkha Samhitha and Shushrutha Samhitha however in the later centuries the whole face of Clinical trials in India has changed with India becoming an internationally acclaimed location for cheaper subjects for performing Clinical Trials. This chapter since discusses the History and evolution of Clinical trials starts off with the Nuremberg Code. The Nuremberg Code involves the convergence of Hippocratic ethics and the protection of human rights into a single code. Further on, this chapter dicusss the Declaration of Helensiki which is a document not only confirming the rights of the subject but also maintains measures to check the obligation of the physician/researcher. Finally this chapter concludes by discussing about Council for International Organizations of Medical Sciences (CIOMS) which are guidelines for all such nations that wanted to draft regulations with consideration to cultural, social and economic condition prevailing in the country.
CHAPTER 4 : JUDICIAL RESPONSE TO CLINICAL TRIALS
Litigation over injuries to research subjects is not new. For at least two decades, occasional cases alleging a failure to obtain informed consent have been brought against various defendants. The doctrine of informed consent dates to the 1914 case of Schloendorff v. New York Hospital Marie admitted to hospital in New York in January 1908 Schloendorff , ” suffering from a disorder of the stomach. ” She accepted a ” review of the ether ” to help identify a lump that was detected. While the patient was under the influence of anesthesia , the surgeon removed a fibroid found during the examination. infection , gangrene and amputation of several fingers would have resulted from the operation.
Mary sued the hospital to maintain the institution responsible for the actions of doctors and nurses employed there . This is the doctrine of respondent superior. There was no claim of responsibility against individual doctors or nurses in this case.
This decision often cited and misunderstood gives a fascinating view of hospital care in the United States , as it existed in the early 20th century. The easy – paid $ 7 per week for care , the poor paid nothing . The decision describes the role of the nurse in patient advice of acerca surgery provided by physicians Marie continued Schloendorff hospital in New York, alleging that doctors performed surgery, it employed. Unlike his express order. The judge asked the defendant hospital to rule on legal grounds, even if the allegations were true , Mary lost. The judge accepted that a directed verdict and included in the income of the defendant hospital . The intermediate-level court , the Appellate Division , First Department , upheld the verdict directed by the trial judge . Schl??ndorff v New York Hospital , 149 App Div 915 ( 1 March 1912 ) .
The case was then brought before the Supreme Court of the State of New York, the Court of Appeal . To select the case, the court had to determine the responsibility of a non-profit hospital for acts performed by doctors and nurses employed drank . Proposed two theories supporting the finding that the hospital was immune from liability for damages suffered by the patient. One theory was that the patient has declined to pursue for the treatment of neglect when the patient turned for help to a charity ( charitable immunity ) . In rejecting this , Cardozo J. summarized the state of the common law relating to consent to surgery
The Court of Appeal held that a hospital could not be held responsible for acts of its employees for physicians . The Court of Appeal of New York has since rejected the “rule Schl??ndorff ” and felt that the principles of respondeat superior should be applied to make the hospital liable for the negligence of the doctors and nurses it employs. V Bing. Thunig , 143 NE2d 3, 9 ( 1957)., in which New York’s highest court articulated a patient’s right to full information before undergoing treatment. Informed consent litigation relating to medical treatment blossomed in the 1960s and 1970s , when courts in many jurisdictions decided to replace a profession based disclosure standard with a patient-based standard. In other words, jurors were asked to judge claims not according to what information physicians thought a patient reasonably should be given but rather according to what the patient might reasonably expect to be told . Claims alleging a negligent failure to obtain informed consent continue to day as a fairly common species of medical malpractice litigation. These claims have been easily transplanted to the research context. The typical allegation is that the research subject was not given sufficient information to permit meaningful consent to research participation. For example, in Berman v. Fred Hutchinson Cancer Center , The plaintiffs in the case filed their complaints after a series of articles in the Seattle Times problems with certain cancer studies at the Fred Hutchinson Cancer Center exposed. Applicants are cancer patients who died received a protocol known as 126 experimental treatment. The plaintiffs alleged that the subjects were not adequately informed about the risks of treatment or financial interests related to the experiences of cancer treatment at the Fred Hutchinson Cancer Center . They also alleged . Researchers have not reported appropriately the deaths and did not make changes to the consent forms. The plaintiffs in Wright, like Robertson also said that the search had violated the right to be treated with dignity grounded in international codes of ethics and due process clause of the 14th amendment . The plaintiffs later amended their complaint and disapproved any claim based on international codes of ethics. The federal district court in Seattle dismissed the plaintiffs’ request that the search violated their right to due process . The court contrasted the consent process in the case of the consent process in D Cincinnati . In Wright, the court found that subjects gave their consent to treatment that could benefit them. Cincinnati case , Since the late 1940s to the 1980s , the U.S. government has conducted over 4000 experiments secret radiation exposure on military personnel and civilians. The official reason for the conduct of these experiments was to gain a better understanding of radiation on human beings to better prepare for a possible nuclear attack on the Soviet Union or to inflict maximum damage on an aggressor country. In 1994 , President Clinton declassified thousands of documents relating to these experiences and established the Advisory Committee on human radiation experiments to study the ethical and legal problems with experiments. The Committee noted that in most experiments, subjects did not consent to procedures or suffered harm important. The secret experiments generated dozens of trials . Some were settled for millions of dollars and others were rejected on the grounds that the government had legal immunity under the Federal Tort Claims Act, which provides the legal protection of the United States government workers conduct discretionary powers under government . However, another federal law allows plaintiffs to prosecute government officials if those officials deny the rights protected by the Constitution or federal statutes Case Cincinnati , the plaintiffs successfully argued that irradiation experiments by doctors on 88 patients the University of Cincinnati from 1960 to 1972 violated the due process rights protected by the 14th Amendment to the Constitution. Patients who had inoperable cancer with an average life expectancy of two years, received no radiation exposure – therapeutic without their consent. The Federal Court found that these experiences have violated the rights of plaintiffs to avoid bodily invasive integrity . The legal significance of this case is that it establishes a method to successfully prosecute government agencies that have Fonn legal immunity under state or federal law. Many organizations and institutions such as the NIH , the Environmental Protection Agency (EPA ), the Department of Defense ( DOD ), the Veterans Administration (VA) governmental different colleges of the state universities , hospitals and clinics to conduct research on human subjects. Although most organizations have some type of legal immunity under state or federal law, they will still be responsible for research on human subjects that violates the rights protected by the Constitution or federal statutes . subjects were not given their consent for any procedure that gave them no advantage. The Court also concluded that federal research regulations do not give rise to federal rights protected by the Constitution or federal statutes On April 9, 2004, a jury found that the Fred Hutchinson Cancer Center had not been negligent in death of four of five subjects. He gave $ 1 million to the family of a deceased subjects. Although the jury found that there were problems with the consent process , it was determined that a reasonably prudent person would have included in the study of cancer , even if he or she had more information about the risks and financial interests brought by the husband of a patient with breast cancer who died in a chemotherapy trial, the plaintiff alleged that his wife would not have agreed to participate in the trial if she had known that a drug intended to prevent lethal side effects from the chemotherapy was not available in intravenous form Contrary to what doctors allegedly told her, the drug was available only in tablet form, and she died after vomiting the pills. In August 2002, the trial court ruled for the plaintiff on this claim .
It is perhaps obvious for most health care professionals that liability will arise where research is conducted without informed consent from the subjects (or their substitute decision makers) being obtained. What may be less known is that it appears from existing case law as though the standard for obtaining informed consent in the research context is higher than in the therapeutic context. The other interesting development has been that it appears that courts may be willing to hold a hospital responsible where the informed consent form approved by the REC of the Hospital is determined to be less than adequate.
In the Weiss case for example, the Quebec Superior Court found that the duty to inform in matters relating to purely scientific experimentation is the most exacting possible and includes the disclosure of all known risks including those which are rare or remote, especially if they may entail grave consequences. This would suggest a standard that is higher than the standard for consent to treatment which requires that only material risks be disclosed. In addition, the court in Weiss found that the hospital was liable and attributed some of that liability to the fact that the hospital’s REC failed to ensure that the consent form used for the research was appropriate .
Government Initiated Legal Proceedings.
In addition to being sued by research subjects who allege being injured by their participation in a study, there is also a possibility that the researchers and institutions involved in health research involving human subjects will face legal battles with governmental bodies/agencies with jurisdiction over research. In the U.S., such bodies have imposed drastic sanctions where there is evidence of research misconduct. The defendants in the Gelsinger case Gelsinger v. University Hospital of Pennsylvania ( 1999). In this case, that was settled out of court for an undisclosed amount of money, succeed Jesse Gelsinger and his family sued the University of Pennsylvania, Genovo , Inc., Children’s Hospital of Pennsylvania, National Children’s Medical Center , James Wilson, Steven Raper, Mark Batshaw , William Kelley, and Arthur Caplan for a variety of claims, including wrongful death , product liability , assault and battery , lack of informed consent negligence and intentional infliction of emotional distress , and fraudulent misrepresentation. Gelsinger died after a severe immune reaction to the infusion of adenovirus vector in the liver. Gelsinger suffered from ornithine transcarbamylase deficiency ( OTD ), a rare metabolic disease . Although most patients with OTD die in infancy , Gelsinger was able to manage the disease through an iris drugs and low-protein diet . Patients OTD lack a functional copy of the necessary for the manufacture of the ornithine transcarbamylase gene , an important liver enzyme . The unfortunate experience tried to use the vector to transfer copies of the gene in the liver Gelsinger . If the experiment had worked , Gelsinger may not need medication or special diet to control metabolic problems. However, the purpose of the experiment was to test the safety of the procedure , and not to assess its effectiveness. The experience had a very low chance of benefit Gelsinger . Voluntary Gelsinger to “experience because he thought it was not very risky and it is hoped that the experience would help researchers develop a treatment for the disease. The experiment was a Phase I: Gelsinger was the first human subject to receive the Vector adenovirus The lawsuit alleges that Gelsinger was not adequately informed about the risks to the experiments and interests financial researchers and the University of Pennsylvania. Wilson and the university both shares owned Genovo , a company founded by Wilson. Genovo also paid $ 4 million per year to the university to support research on gene therapy. Wilson holds several patents on gene therapies , methods . Although the case was settled out of court and does not establish legal precedent , therefore , there was a ” significant impact on ethical and regulatory aspects of clinical research . In the wake of the case, the media national once focused its attention on human research , lawyers began to file more lawsuits related to human research , government agencies issued new guidelines on conflicts of interest ( CLO) and increased their oversight of research and professional organizations and journals also developed or revised policies Col .not only settled with the plaintiffs, they also settled with the U.S. government in a separate civil action commenced by the government on the basis of breach of the False Claims Act The settlement resulted in a total of over one million dollars in payments to the government by the institutions involved in the research as well as restrictive controls being placed on three investigators involved in the research with respect to their future clinical research activities .
Law suits based on Principles of International Human Rights
In the case of Abdullahi v. Pfizer, Inc. , Pfizer was the defendant in a lawsuit that alleged that it improperly administered an experimental antibiotic to children in Nigeria during an outbreak of bacterial meningitis, measles and cholera in Kano, Nigeria. The guardians of certain of those children instituted an action in the Southern District of New York, alleging violations of the Nuremberg Code, the Declaration of Helsinki. Article 7 of the International Covenant on Civil and Political Rights, U.S. Food and Drug Administration’s regulations and other norms of international law. They also asserted that the court had jurisdiction over the matter under the Alien Torts Claim Act. This case was sent back to the District Court to determine whether the U.S. or Nigeria is the appropriate forum to hear the case Even In the Robertson case, several of the causes of action were derived from international human rights law rather than standard medical malpractice or tort law, including allegations of breaching the right to be treated with dignity, citing the Nuremberg Code and Declaration of Helsinki concerning biomedical research.
The New Era of Medical Claims
Enterprising plaintiffs’ attorneys have turned to a daunting array of legal doctrines in framing lawsuits against those who perform and oversee research. A lawsuit that 20 years ago would have been brought as a routine informed consent claim may today include allegations of defective products, fraud, negligent conduct and monitoring of the research, intentional infliction of emotional distress, breach of patients’ rights protected by state law, violation of federal regulations, and violation of constitutional rights. A popular approach in lawsuits alleging injuries in pharmaceutical trials is to combine the traditional informed consent claim with product liability claims against the drug manufacturer .For example, the mother of an infant who died in a trial of the heartburn drug Propulsid filed suit against the Children’s Hospital of Pittsburgh and Janssen Pharmaceuticals, alleging that she was not informed of risks and adverse events associated with Propulsid and that the drug was defectively designed . This legal tactic recycles was once used in some of the earliest human subjects’ litigation, claims brought by women whose infants were injured by diethylstilbestrol . Three other recent cases illustrate even greater legal creativity in the drafting of claims. The case of Gelsinger v. University of Pennsylvania Hospital arose from the death of 18-year-old Jesse Gelsinger in ‘a phase I gene therapy trial , The Gelsinger family coupled the usual informed consent claim with a product liability claim, and then went further. They alleged that the investigators had committed fraud by not revealing that previous subjects enrolled in the protocol had died and that the principal investigator had a financial relationship with the sponsoring biotechnology companies The parties reached a confidential settlement in November 2000. Robertson v. Oklahoma , This case is due to problems with a vaccine against melanoma prepared by researchers at the University of Oklahoma Health Sciences Center. The university has suspended “his study after an internal audit revealed problems affecting patient safety, including the lack of control over the preparation of vaccine. The audit also revealed that the researchers had not told their subjects or the FDA about these problems. A whistleblower informed Office of Human Research Protections [OHRP ] about these problems. OHRP that time has suspended all research funded by the federal government on the campus of Oklahoma, found major violations of federal regulations vaccine study and found the IRB was negligent in its supervision of research and monitoring protocols. The case is legally important because it is considered the first case in American law that the names of individual IRB members as defendants. Other cases were appointed to the Board, collectively, as a defendant the case is also important because a federal district court refused to recognize the right to be treated with dignity based on international ethical standards such as the Nuremberg Code. The plaintiffs had argued that the search violated their right to be treated with dignity, but the court rejected this claim as a vague unsupported by federal law. The Federal Court found that the allegations of the accused were better interpreted as actions in tort liability state, not actions104 federal civil rights . In their complaint, the plaintiffs also allege that the investigators did not fully inform the risks of the vaccine and that investigators deformed as a cure against cancer. They also said that investigators enrolled ineligible subjects and did not adequately monitored security. In July 2002 some of the defendants reached a settlement with the plaintiffs . a state court case that grew out of a federal suit dismissed for lack of jurisdiction54, involved injuries to subjects in a phase III melanoma vaccine trial at the University of Oklahoma Health Sciences Center at Tulsa. The plaintiffs made three main allegations. First, they alleged a lack of informed consent based on the investigators’ failure to reveal that the vaccine had not been subjected to animal studies, their failure to disclose all the relevant risks, and their misrepresentation of the vaccine as a potential cure for cancer. Second, they claimed that the trial itself was negligently run essentially a claim of investigator “malpractice” in the conduct of research because the investigators enrolled ineligible patients and failed to monitor the subjects’ health appropriately. Third, they alleged that investigators fraudulently misrepresented the purpose, risks, and benefits of the study. Some of the defendants reached a settlement with the plaintiffs in July 2002 The case of Wright v. Fred Hutchinson Cancer Center is another interesting cocktail of legal claims; it incorporates the kind of “research malpractice” and fraud claims used in Robertson, includes conflict of interest allegations like those in Gelsinger, and adds several new theories. In this case, subjects in a trial to prevent graft failure in patients undergoing bone marrow transplantation alleged that the investigators used misleading consent forms and failed to disclose various conflicts of interest. The plaintiffs also claimed that the investigators failed to report deaths appropriately to the IRB and failed to update consent forms as required by the IRB, in essence alleging negligent conduct of the trial. In addition, the plaintiffs in Wright posited a “breach of the right to be treated with dignity” under the due process clause of the 14th Amendment to the U.S. Constitution, arguing that international conventions such as the Nuremburg Code and the Declaration of Helsinki substantiated this right. The same groups of attorneys have taken these groundbreaking constitutional and international law claims even further in other cases, although it is too early to judge their legal sway. The trial court in Wright dismissed the constitutional and international law claims made in that case, while a New York district court allowed similar claims to proceed in a case involving a drug trial in Nigeria. The fraud claims in cases like Wright are of particular interest because they give the plaintiffs’ attorney significant leverage in court. An allegation of fraud is likely to powerfully affect jurors. While the public may be becoming more cynical about business ethics in the wake of several major corporate scandals, the public is used to thinking of researchers as committed to new knowledge and the welfare of research subjects rather than the pursuit of profits that might come from a successful trial. Such a claim, casting investigators as intentionally leading subjects into danger for their own financial gain, can alter the entire tenor of a case. Fraud allegations also open the door for enormous damages awards. In tort law, damages may have three components: compensation for economic losses, such as lost wages; damages for pain and suffering; and “punitive” damages. The punitive damages component, which is intended to punish especially blameworthy defendants, usually accounts for a substantial portion of the multimillion dollar personal injury verdicts that attract media attention. In actuality, punitive damages are rare , They occur in less than 1.5% of medical malpractice verdicts and approximately 5% of plaintiff trial wins . However, punitive damage awards are exceptionally common among fraud claims, occurring in about one fourth of verdicts for the plaintiff. Hence, research subjects who bring successful fraud claims stand a good chance of receiving very large damages awards. This makes research-related litigation very attractive to plaintiffs’ attorneys who work on a contingency-fee basis .
THE LEGAL BAISIS OF COMPENSATION FOR CLINICAL TRIALS
Research subjects have a right to seek redress for their injuries through the legal system. If a subject is injured during research, he of she may be able to bring a lawsuit against many different parties implicated in the injury, including: researchers; research staff, such as nurses or patient advocates; institutions, such as universities, medical centers, or hospitals; sponsors, such as pharmaceutical or medical device companies; and even institutional review board members. Most of the causes of action brought against defendants in research litigation have involved various torts, such as battery, negligence, fraud, misrepresentation, conversion, unjust enrichment, breach of fiduciary duty, violation of informed consent, products liability, intentional infliction of emotional distress, and wrongful death. Some lawsuits against government agencies or institutions have sought compensation for civil rights violations, and a small number of cases have addressed breaches of contractual duties . Although there are no data on the success rate of human research lawsuits, it may be reasonable to extrapolate from data on medical malpractice cases, which are settled out of court 96% of the time When plaintiffs manage to get a verdict at trial, they win less than 30% of the time A malpractice claim is filed in only one out of eight cases of medical error that result in an injury. If these percentages also hold for human research lawsuits, then most subjects who have research-related injuries will not bring a lawsuit. When subjects do bring litigation, their cases will be settled out of court most of the time. And if a case ever goes all the way through a trial, the plaintiff will lose most of the time. Even though the chances are probably very small that subjects will bring litigation for their injuries. the liability risks still are a cause for great concern. Lawsuits can be very stressful. cost millions of dollars in legal fees. and continue for many years until they finally are dropped. settled out of court, or adjudicated. It is quite reasonable, therefore, for researchers. institutions, and sponsors to take steps to avoid litigation or minimize its impact. A plan for providing subjects with compensation for research related injuries can help lower the risks of litigation by encouraging subjects to seek help through the plan, instead of seeking legal redress. It makes good sense, from a legal risk management perspective, to purchase insurance or set aside funds to compensate subjects for research-related injuries and develop a system for administering the plan . Although this legal risk management perspective provides a sound justification for adopting a compensation plan, it also creates some ethical and legal problems. The first problem arises in trying to limit legal liability associated with developing a plan. Suppose that a research sponsor wants to offer to pay for medical treatment for injuries caused by research participation, but nothing more. Someone who develops the language used to communicate the plan to subjects may be tempted to try to minimize legal risks by requiring potential plaintiffs (in other words, research subjects) to forego some legal rights to participate in research. The current system of compensation for research related injuries is unjust because it allows for inconsistent treatment of human subjects and does not require research institutions or sponsors to compensate subjects for research-related injuries. To correct the injustices in this system, the government should revise the human research regulations to set a minimum standard for compensation plans. In addition, organizations that promote ethics in clinical research should develop standards for compensation plans . Death of volunteers at Johns Hopkins in 2001: Volunteer , Ellen Roche , died on 2 June , a month after inhaling unapproved drug in a study to examine the causes of asthma medication. The study for which this voluntary person was funded by a grant from the NIH with the title “lung inflation and Airways Hyper responsiveness. ” The study was a physiological basic test using inhaled hexamethonium for how significant lung function , which protect the airways to narrowly , works. This feature plays a critical role in the development of asthma. Her lungs were destroyed, apparently by the chemical she inhaled hexamethonium . She was 24 , was a volunteer employee (technician at Johns Hopkins) incomplete search by researcher claims no complaint , but OHRP actions led to the law of the State on the activities of the IRB
In Quinn V. Abiomed Inc., et all In this case, the estate of James Quinn and his widow Irene Quinn suing Abiomed , Tenet Healthcare, Drexel University, Hahnemann University Hospital, and David Cassarett of negligence , lack of informed consent , battery, intentional infliction of neglect and distress emotional , fraud, product liability , survival measures , and loss of consortium. In September 2001, James Quinn, aged 51 was diagnosed with congestive heart failure . His doctor said he had less than six months to live. Abiomed is preparing to conduct clinical trials on its new artificial heart. James Quinn enrolled in the trial, and on November 5-2001 , he received a new heart manufactured by Abiomed . Over the next 10 months, Quinn had a series of medical complications of the experimental device, including stroke, breathing difficulties , bleeding, partial paralysis , vision loss , pneumonia and severe pain. He had limited mobility and spent much of his time in the hospital. Quinn died August 25, 2002 , following a massive stroke . On October 16, 2002, counsel for the Quinn’s, Alan Milstein, filed a complaint against the defendants. Most of the claims in this case focuses on the alleged problems with the consent process. According to the plaintiffs’ complaint, Quinn has not been given enough information about the risks of the device and did not say he was participating in an experiment. The informed consent document signed Quinn described this experience as a “clinical initial feasibility study .” The complaint also alleges that Quinn was particularly vulnerable because it was near the end of life. In an interview, Milstein says Quinn succumbed to the therapeutic misconception, well – materials phenomena in which humans believe that the main study objective is to provide treatment, when in fact the main objective of the study is to test a device or a drug . Persons error medical experiments for medical treatment , even when they are told that their participation in research that can offer them no benefit . A unique feature of this case is that it names a patient advocate , David Cassarett that the defendant . Cassarett of a biomed was paid by Quinn to provide information and help them decide to participate in the experiment.
In Moore V. The Regents of University of California Applicant , John Moore went to UCLA Medical Center after being diagnosed with leukemia hairy cell . Doctor Moore, David Golde, was diagnosed with cancer in 1976 Moore at the University of California at Los Angeles (UCLA) Medical Center and recommended that Moore had a splenectomy. Moore signed Form of written consent authorizing the procedure. Golde and his colleague, Shirley Quinn, had made plans to study the rate of Moore: That was an overproduction of proteins precious immune system. Defendant told the plaintiff follow-up care could be performed at UCLA Medical Center and the applicant travel expenses incurred. From 1976 to 1983, Golde asked Moore to return to the UCLA Medical Center from his home in Seattle to provide additional samples of blood, skin, bone marrow , and Spleen . Cells applicants were very helpful to the defendant and were used to help develop and patent a cell line that was sold to Genetics Institute and Sandoz Pharmaceuticals, also accused. In 1979, Golde and Quinn had (established a cell line of T cells in the spleen of Moore, and in 1981 they applied for a patent was awarded in 1984. Golde and Quinn assigned their patent rights to the University of California the time the patent was issued, the market for cell line was estimated at $ 3 billion. Though Moore had consented to the surgery, I had not consented to the use of the tissue of conduct research or develop commercial products . ‘ve also not been informed of the financial interests of Golde , Quinn, or university in the research.88 Moore sued the researchers, universities, and private companies when yo have discovered that they were patenting a cell line developed from his spleen cancer . plaintiff filed a complaint raising thirteen causes of action, including conversion , among others, and lack of consent . Moore continued the researchers, university, and private companies for conversion ( a tort of unlawful interference with personal property) , breach of fiduciary duty, and lack of infonned consent. The trial court sustained a demurrer to the count of conversion and dismissed the complaint without ruling stating that the other issues raised were insufficient conversion repetition of the allegation. The case eventually reached the Supreme Court of California . To prove his conversion request , Moore needed to show that the cells were his property, even if they were removed from his body. The Supreme Court of California has not confirmed the conversion of Moore ‘s claim on the ground that Moore did not have a right of ownership in large cells eleven They were removed from his body, but the researchers had a property right in the cells because they help to develop a patentable invention. If Moore had succeeded in proving its conversion application, I would not have had the right to share the benefits of its cell line. But the Court held in Moore , on behalf of his other claims. It’s Golde He stood as doctor Moore, the legal obligation to declare foreign interests to the health of Moore, including economic interests and the interests of research , Quinn affect his professional judgment . The question before the California Supreme Court question is what defendant physician breached the fiduciary duty and failed to obtain the consent of infonned patient. The Court agreed with the applicant. The Court states that a person of adult age and his mind has the right, in the exercise of control over his own body , to determine whether or not to submit to a legitimate medical treatment patients consent a treatment to be effective , must be informed to seek the consent of the patients consent , a physician has a fiduciary duty to disclose all documents to patient decision . In essence , the physician must have staff or any financial interest in the patient I could tell the patient the doctor is biased towards having the surgery can be beneficial for a better doctor and the patient said. Since Golde did not inform Moore of his interest in the use of cells in research or development to develop commercial products of its cells , I perfonned Moore on medical procedures without adequate informed consent and violated its fiduciary obligations to Moore. The legal scope of Moore Is that the case clearly establishes that clinical researchers have a legal obligation to declare financial interests and research to their patients / subjects. The Regulations specifically federal research does not address the disclosure of financial and other interests in research .
In Kits V. Sherman Hospital , In this case, the plaintiff, Richard Kus, sued her surgeon, Gordon Vancil , Sherman Hospital , and two companies of medical devices for medical negligence and battery for their failure to obtain informed consent for the implantation of intraocular lenses. Thehospital IRB and FDA had approved informed consent document for the implementation of intraocular lenses, which contained a paragraph describing the experimental nature of the lenses. Kus testified that Vancil have never said that the lenses were under clinical investigation to prove their safety and effectiveness. The consent form signed by Kus was not the same as the form approved by the IRB since the paragraph describing the experiment had been removed. A receptionist for Vancil testified that Vancil ordered paragraph removed informed consent documents for all 43 patients who received the lenses. Shortly after eye surgery Kus , the FDA issued a letter to the IRB Chair of Sherman Hospital stating that the FDA had withdrawn its exemption experimental device for the lens , which means no ‘ other locations lens should take place. The legal significance of this case is that the court has examined the role of the IRB and the hospital to ensure that researchers obtain the legally effective informed consent before starting the procedure . The court ruled that the hospital has a duty to inform the patient of the experimental nature of the surgery , to make the continuous review of research, in accordance with federal research and to ensure that researchers use consent forms approved by the IRB other courts have held that the hospitals do not have a duty to ensure that clinical researchers to obtain informed consent legally effective subjects . Although plaintiff did not continue the IRB , he would have a strong case against the committee because the IRB failed to ensure that Vancil used the consent form was approved.
In Diaz v. Hillsborough County Hospital Authority , This Was Another case settled out of court, so it sets no legal precedent. This was a class action law suit On Behalf Of 383 pregnant women Who Were Subjected to medical experiments,: such as amniocentesis, without Their consent. Researchers used the English consent forms, even though many of the women did not speak English. Some of the women “consented” to the research falling on admission to the hospital for Labor while drowsy or delirious gold. The Plaintiffs Claimed That the Defendants Violated their right to refuse the needed treatment as well as a right to be Treated with dignity. In the settlement, the hospital Agreed to make payments to the Plaintiffs and exchange research procedures ‘
In Grimes Y. Kennedy Krieger Institute In this case, the plaintiffs sued Kennedy Krieger Institute ( KKI ), which is affiliated with Johns Hopkins University , negligence , claiming that the accused did not have to comply with its duty to inform applicants about the levels of hazardous lead and failed to obtain adequate informed consent. KKI was induct a study on methods for reducing lead emissions and families recruitment in the Baltimore area. KKI offered to different types of families reduce lead emissions and concentrations of lead in dust and children’s blood. Although researchers intend to inform applicants about dangerous levels of lead, the plaintiffs allege that they were not timely. The complainants also alleged that they are not sufficiently informed about the risks of the study. KKI presented in a motion for summary judgment of the district court, arguing that plaintiffs had no legal basis for a lawsuit because of negligence KKI had no legal rights to complainants . The district court ruled in favor of KKI, but the Court of Appeal found that KKI had legal duties to applicants on the basis of a contract with the plaintiffs (contained in the informed consent document) and legal rights imposed by the common rule. The Court of Appeal remanded the case to the district court for further proceedings consistent with its decision . The court also held that KKI had a duty to warn the parents of subjects of dangerous levels of lead and obligation to obtain legally valid informed consent of the parents. The Court of Appeal also considered the legal standards to include children in research that offers no prospect of direct benefit (also known as non-therapeutic research). The court first concluded that children cannot participate in this type of research unless it is expected to present no risk to subjects. However, the court later changed its mind on the issue of pediatric research since its initial decision was not consistent with federal regulations , which allow children to participate in non- beneficial research which presents minimal risk or only a slight increase in minimum risk Grimes is a legally important case because it is a rare case to discuss pediatric research and because it also addresses the functions that researchers to research subjects when there is no therapeutic relationship as a medical patient. Other cases have firmly established that researchers have legal obligations to their subjects when their subjects are also patients. The court ruled that Grimes researchers have legal obligations to subjects in the absence of a doctor-patient relationship. The relationship between the researcher and the subject is a special relationship which may give rise to legal obligations.
In Kernkev Menninger Clinic In this case, The parents of the subject of the research sued “clinical and pharmaceutical Aventis continued after the death of the subject given experimental drug for schizophrenia and affective disorders schizophrenia. It was alleged that the consent form was flawed. The Court has used “intermediate doctrine competent” to absolve Aventis right to complainants. It was held that Aventis had given security relevant information and the effectiveness of Menninger Clinic in the brochure of the drug.
In Robertson V. McGee This case is due to the problems with a vaccine against melanoma prepared by researchers at the University of Oklahoma Health Sciences Center. The university had suspended “his study after an internal audit revealed problems affecting patient safety , including the lack of control over the preparation of vaccine. The audit also revealed that the researchers had not told their subjects or the FDA about these problems. A whistleblower informed Office of Human Research Protections [ OHRP ] about these problems. OHRP. that time had suspended all research funded by the federal government on the campus of Oklahoma , found major violations of federal regulations vaccine study and found the IRB was negligent in its supervision of research and continued evaluation protocols. The case is legally important because it is considered the first case in American law in which the names of individual IRB members were brought up as defendants . Other cases were appointed to the Board, collectively, as a defendant The case is also important because a federal district court refused to recognize the right to be treated with dignity based on international ethical standards such as the Nuremberg Code. The plaintiffs had argued that the search violated their right to be treated with dignity, but the court rejected this claim as vague unsupported by federal law. The Federal Court found that the allegations of the accused were better interpreted as actions in tort liability by state not actions federal civil rights . In their complaint, the plaintiffs also allege that the investigators did not fully inform the risks of the vaccine and that investigators deformed as a cure against cancer. They also said that investigators enrolled ineligible subjects and did not adequately monitored security. In July 2002 some of the defendants reached a settlement with the plaintiffs
In Greenberg v. Miami Children’s Hospital (2003) . In this case it centered on a dispute over genetic testing for the gene for Canavan disease that erupted in the late 1990s trial. The plaintiffs included parents of children with Canavan disease and non-profit organizations that provided funding and support for research for Canavan gene . The defendants included Reuban Matalon , a physician and researcher in genetics Hospital Miami Children’s Hospital (MCH ) and the Institute for Children’s Research Hospital Miami . Canavan disease is a rare genetic disease that affects people descended from Ashkenazi Jews . Approximately 2.3 % of this ethnic group gene carriers . The disease is a fatal , incurable , degenerative brain disease that occurs when a child is three to six months . In 1987 , Daniel Greenberg, one of the complainants , asked if he would be interested in Matalon help develop a test to identify disease genes Canavan provide information on the carriers to help in the decisions concerning reproduction. Matalon , who at that time was at the University of Illinois at Chicago, has agreed to help find these genes. Greenberg and Chapter of the National Tay- Sachs and Allied Diseases Association ( NTSAD ) Chicago started communicating with Canavan families to ask for samples of fabrics, genealogical information , and financial support. Greenberg and NTSAD formed a national registry Canavan and started the Foundation Canavan . In 1990, Matalon moved to MCR . In 1993, Matalon isolated the gene for Canavan . In 1994, Matalon has filed a patent on the Canavan gene, and in 1997 he received the patent, which he entrusted to the MCH. The patent has the right to restrict other MCH to conduct genetic testing for Canavan gene ll2 . MCH considered a gift of his patent rights to the public and the above charges , but he fears that laboratories would not know the test. In 1998, MCH began exercising its intellectual property related to gene Canavan and threatened to sue organizations that offered testing for gene patent infringement. MCH has entered into licensing agreements with certain organizations and royalties charged $ 12.50 per test. MCH used license fee money to offset the cost of test development and pay for pUblicityll3 . Although SMI had established a very modest price for testing, members of the Canavan community were convinced they should have free access to the event, especially since they have contributed to its development. On October 30, 2000 , the plaintiffs sued the defendants in federal court for a variety of claims, including lack of informed consent , breach of fiduciary duty , fraud , conversion and unjust enrichment . The court rejected all these allegations , except unjust enrichment .
The court followed Moore in rejecting the application for conversion. The court ruled that the plaintiffs did not have a property right in their donated tissue. However, the court did not reject the application of unjust enrichment, because the plaintiffs had invested time and effort to help identify the gene , and therefore deserved to share the benefits of discovery. The most interesting and controversial decision of the Court regards the allegations concerning informed consent and fiduciary obligations. The plaintiffs argued that the defendants did not disclose financial interests, i.e. the interests of the patentability of the gene when they donated tissue. The complainants contended, citing Moore and Grimes , that the defendants have a duty to disclose those interests. However, the court rejected this argument , saying that the duty of a researcher to disclose the financial interest has a strong legal precedent , and the extension of the requirement of informed consent economic interests would have a negative impact on research. The plaintiffs also argued, citing Moore and Grimes , that the defendants had fiduciary duties to the plaintiffs, which involves legal obligations to declare financial interests. However, the court also rejected this argument by distinguishing between therapeutic and non-therapeutic research. When a therapeutic relationship, such as doctor-patient exists between researchers and subjects, researchers have fiduciary duties towards the subjects. When there is no such relationship, the researchers do not have these legal obligations, the court said. Since the defendants did not have a therapeutic relationship with the plaintiffs, they had no fiduciary duties. The court also rejected the claim made by Grimes that there is a special relationship between the researcher and the object Thus, courts Grimes and Greenberg have very different decisions on the nature of the relationship between the researcher and the object research. They both agree that when the researcher is a doctor and the subject is a patient, the researcher had a fiduciary duty to the object, but they disagree on the nature of the relationship when the researcher n ‘ is not a physician (or other health care provider) and the subject is not a patient. Since the opinion of Grimes was delivered by an appellate court of the State, and Greenberg opinion was issued by a federal district court, the court Greenberg is not required to follow the Grimes court.
In Suthers, et al. v. Amgen, Inc., In this case, Milstein recently filed with the Federal Court of the District of Manhattan, Robert Martin Suthers and Niwana Amgen continues to stop a clinical trial of the drug glial cell line – derived neurotrophic factor experimental (GDNF ) , used to treat Parkinson’s disease. The company has stopped the trial on the ground that it had evidence that the drug is ineffective and dangerous. According to Amgen, an analysis of data from human studies have shown that GDNF is not more effective than placebo, and animal studies have shown that high doses of GDNF can cause brain damage. The prosecution contends that Amgen has an obligation to continue to provide the drug to patients because he had a contract with patients through the consent process, to conduct the trial. The two subjects underwent surgery to enable them to receive the drug, that has been delivered to the brain through tubing below their skin . A victim of an attack patient after surgery and had to have her tube re- implanted. The lawsuit alleges that the company promised to continue to provide the drug to subjects as he helped. Suthers said the drug helped to walk up to two miles on a regular basis , and now he can walk just a few blocks away. The case is unique in so far as it is a complaint against a company to stop a clinical trial to protect the so-called research subjects against evil .
Clinical Trial Litigation Trends in India
It has become increasingly difficult to test drugs in Western countries, with their strict regulations, elaborate safety and compensation requirements, and small populations, all of which make the recruitment of research subjects slow and expensive. Consequently, many research-based companies are now outsourcing some of their trials to Third World countries such as China, Indonesia, Thailand, and India. India is a particularly attractive destination for such trials because of its genetically diverse population of more than 1 billion people who have not been exposed to many medications but have myriad diseases, ranging from tropical infections to degenerative disorders. Virtually all Indian doctors speak English, and many have acquired postgraduate qualifications abroad, primarily in Britain or the United States. Added to these attractions are cheap labour and low infrastructure costs, which can reduce expenditures for clinical trials by as much as 60 percent However, even from the viewpoint of foreign drug companies, there are some major drawbacks to working in India. Sponsors do not have exclusive rights to the clinical data they generate, because trial reports are in the public domain, manufacturers of generic drugs can use the data to obtain regulatory approval of their own versions of a drug. Furthermore, the Drugs Controller General of India (DCGI) the equivalent of the U.S. Food and Drug Administration (FDA) is understaffed and lacks the expertise to evaluate protocols. Currently, the technical staff consists of just three pharmacists, including the controller, and not one medically qualified doctor. As a result, persistent follow-up, including personal visits to the DCGI, is required in order to push an application for a trial forward. In addition, although the country has more than half a million practicing doctors, fewer than 200 investigators have been trained in good clinical practice. Among some 14,000 general hospitals, no more than 150 have the adequate infrastructure to conduct trials. and there are fewer than a dozen pathology laboratories that meet the criteria for compliance with good laboratory practice. Only about half of the large hospitals have institutional review boards, and even these boards have not yet formulated standard operating procedures and they, too, often lack the expertise with which to evaluate protocols. Information about conflicts of interest is neither sought nor voluntarily provided by in,vestigators. Given the sorry state of the apparatus for reviewing proposals, the greatest concern about clinical trials in India, from the vantage point of both Indians and ethicists, is illegal and unethical trials, a number of which have attracted adverse coverage in the media in recent years. In 2002, two new chemical entities, called M4 Nand G4 N. that had been discovered in the United States were tested in 26 patients with oral cancer at the government-run Regional Cancer Center in Kerala. In the same year, self-styled researchers working in their own clinics formulated “vaginal pellets” of erythromycin and tried them as contraceptive agents in more than 790 poor, illiterate, rural women in West Bengal. In 2003, letrozole, an anticancer drug, was tested in more than 430 young women at a dozen private clinics to find out whether it promoted ovulation. All these trials took place without regulatory approval. These studies were conducted by Indian organizations, but in the past, Western pharmaceutical companies have conducted similarly unethical trials Moreover, the sponsors of many such trials engage in practices that are currently legal yet ethically dubious. They have been known to offer financial inducements to participants such as paying illiterate blue-collar workers more per month to participate in a trial than they earn at their jobs and enticing subjects by providing medication that is worth more than their annual salary. Widespread illiteracy makes it particularly easy to sidestep the standard methods of obtaining informed consent. Investigators frequently enroll patients in trials as if their participation were a necessary next step in their care. And no protocol we have ever seen has promised to continue to supply the studied medication free of charge after completion of the trial, if it is found to be beneficial.
On September 24, the Supreme Court admitted for hearing a public interest litigation filed by a non-governmental organization (NGO), Aadar Destitute and Old People Home of Hyderabad, alleging that certain pharmaceutical companies are conducting clinical trials of genetically engineered (GE) drugs on human beings without obtaining permission from the Genetic Engineering Approval Committee (GEAC). The NGO further alleged that the tests were being conducted on poor and destitute people.
The Central Government, in its counter-petition, conceded that some patients had died during clinical trials of certain GE drugs and argued that the mortality rate was well within international norms. The Supreme Court declined to pass an interim order staying all such trials without prior authorization and said the matter had to be heard before any order could be passed as human lives were involved.
According to Monthly Index of Medical Specialties in India, an independent pharmaceuticals journal, more than 400 women who had been trying in vain to conceive were enrolled in 2003 without their knowledge or consent to take part in clinical trials across India to see if a drug called Letrozole induced ovulation. Letrozole used in India was copied (with permission) by Sun Pharmaceuticals, a large Indian generic drug company, from a patented product of the same name of Novartis, which the multinational drug maker introduced globally for solely treating breast cancer and not for any other use in any country, including India. A complaint on the Letrozolexxxiv case, too, was filed in die Supreme Court by yet another Delhi based NGO. And in 2001, another trial that made headlines involved the clinical trial of nordihydroguairetic acid, a chemical with anti??cancer properties that was tested by a regional cancer-treatment center (RCC) in the Indian state of Kerala for a US-based researcher then associated with Johns Hopkins Hospital in the United States. The drug was allegedly tried on 26 unsuspecting cancer patients, two of whom died. Subsequently, a 60-year-old woman was again included for a trial for which the RCC provided five doses of the experimental drug, worth Rs 10,000 (about US$200), free. The woman’s condition turned critical as well before the fifth dose, although she escaped death.
These instances indicate that in the absence of adequate regulations and proper laws, a developing country eager to cash in on the opportunities of globalization can be used for indulging in rash and risky practices. A recent survey of 200 health researchers globally that was commissioned by the former US National Bioethics Advisory Commission and published in February’s edition of the Journal of Medical Ethics revealed that a quarter of clinical trials conducted in developing countries did not undergo ethical review. However, in India particularly, unethical and illegal clinical trials are most rampant and are conducted without fear because, there is no law to safeguard the interests of volunteers, while regulatory authorities, “by design or default”, fail to take action against such trials. There is a need to bring Indian regulatory system in line with international agencies. It goes without saying that effective regulations are necessary to govern the conduct of clinical trials on human beings in India. This issue gains even more importance in the light of the fact that India is increasingly becoming favored destination for Pharmaceutical companies to carry out clinical trials, due to a variety of reasons such as availability of a rich genetic pool, low cost for obtaining volunteers, lack of stringent regulations etc. Under our law, permission should be obtained from the DGCI before the commencement of clinical trials, but there is little regulatory control after that. The regulations set out in Schedule Y to the drugs and cosmetics rules are rudimentary and do not specifically regulate the different aspects of clinical trials. Though the guidelines brought out by the ICMR are detailed, they lack statutory backing. More importantly, in the absence of an enforcement mechanism, there is no way of ensuring that Sponsors and Investigators observe ethical standards and guidelines while conducting trials on human beings. Such an enforcement mechanism is absolutely necessary in a country like India where the “volunteers” chosen for the trial are often ignorant and lured by economic incentives. Even if an incident of violation of the Rules or Guidelines comes to light, there is no definite remedy that is provided for in the law. In the case of patients subjected to trials at the Regional Cancer Centre in Kerala, the matter was reported to the State Human Rights Commission which has powers to order compensation and also pass orders in the interest of justice. Thereafter, the DGCI also canceled the permission granted to the Centre to carry out the trials. The case only highlights the need to develop appropriate enforcement or redressal mechanisms that would ensure the observance of clinical and ethical standards in the conduct of trials on human beings. The law relating to clinical trials is far from clear and it seems that subjects who participate in clinical trials are the least protected by law in India.